Duration: 5
min

Jeffery Shi
Protein and Antibody Product Marketing
Jeffrey Shi, Head of Protein and Antibody Product Marketing Team of Marketing
Department. He and his team are responsible for customer-centric development of full product
life cycle management for Protein and Antibody, and drive the sustainable development of the
protein antibody business.
Introduction
Throughout humanity's eternal struggle against disease, each medical breakthrough — from the
serendipitous discovery of antibiotics to the precision-guided era of targeted therapies — has rewritten the
rules of modern medicine. Now, bispecific antibodies (BsAbs) are stealing the spotlight, ushering in an
unprecedented revolution in biopharmaceutical innovation. These ingenious molecules, capable of simultaneously
targeting two distinct antigens, are blurring the boundaries of therapeutic possibility, offering renewed hope
to millions battling complex diseases.
The birth of BsAbs is a testament to scientists' deepening mastery of the immune system.
Traditional monoclonal antibodies (mAbs), while revolutionary, act as single-target snipers. BsAbs, however, are
molecular diplomats engineered through genetic wizardry. The integration of two antigen-binding domains within a
single molecule enables precise dual-target engagement through coordinated molecular interactions. Picture this:
one arm latches onto a tumor cell's "death signal," while the other rallies immune cells for a coordinated
strike. This "two-birds-one-stone" strategy has transformed cancer immunotherapy, autoimmune treatments, and
beyond.
At the molecular level, bispecific antibodies (BsAbs) demonstrate remarkable therapeutic potential
through dual-target engagement capabilities. Through ingenious designs, BsAbs ensure that both antigen-binding
domains maintain an optimal spatial conformation, enabling highly efficient target engagement. This structural
innovation has unlocked immense potential for BsAbs in fields such as cancer therapy and autoimmune disease
treatment. However, the precision of BsAbs is not innate. Unlike traditional IgG antibodies, which consist of
two identical heavy chains and two identical light chains, BsAbs naturally possess four(or three) distinct
chains. This complexity introduces a variety of chain mismatch scenarios, significantly hampering
production
efficiency and therapeutic efficacy.
Creating these as complex molecules is no simple endeavor. Unlike traditional IgG antibodies with
their symmetrical two-chain simplicity, BsAbs juggle four(or three) distinct chains — a recipe for
chaos. Early
designs faced a "molecular identity crisis," with mismatched chains scrambling like tangled earphones, crippling
production efficiency and therapeutic potency.
This is where CrossMab shines. Developed in 2011 by Roche’s scientific mavericks as an upgrade to the classic
"knobs-into-holes" (KiH) technique, CrossMab acts as a molecular dating app for antibody chains. It ensures each
light chain pairs exclusively with its destined heavy chain — rarely awkward mismatches. Compared to formats
based on ScFV or Fab constructs, CrossMab technology demonstrates multiple advantages in terms of stability,
developability, and versatility. CrossMab represents a straightforward, clinically validated antibody
engineering solution that enables the correct pairing of heavy and light chains with minimal engineering effort,
utilizing existing antibody pairs. In fact, CrossMab, in combination with KiH technology, has evolved into one
of the most mature, versatile, and widely adopted technologies in both industry and academia.
CrossMab
CrossMab initially incorporates the Knobs-into-Holes (KiH) heterodimerization domain within the Fc
region, followed by the exchange of heavy and light chain domains in one of the antigen-binding fragments (Fab)
of the bispecific antibody, ensuring the correct pairing of each light chain with its cognate heavy chain. The
"crossover" design preserves antigen-binding affinity while creating significant structural asymmetry between
the two arms of the bispecific antibody, thereby suppressing light chain mispairing (Fig. 1 [1]). For
instance,
swapping the CL-VL domain with the CH1-VH domain in one arm results in a new "light" chain 2 composed solely of
heavy chain domains, preventing its assembly with the original heavy chain 1. Similarly, the unmodified original
light chain 1 cannot interact with the new "heavy" chain 2 on the crossover side, as both contain light chain
domains (Fig. 1B [1]). This approach significantly reduces the likelihood of light chain mispairing.
CrossMab
employs three primary methods for heavy and light chain domain exchange: (1) CrossMabFab, which swaps the CL-VL
domain with the CH1-VH domain; (2) CrossMabVH-VL, which exchanges the VL and VH domains; and (3) CrossMabCH1-CL,
which swaps the CH1 and CL domains (Fig. 1C,D,E [1]).
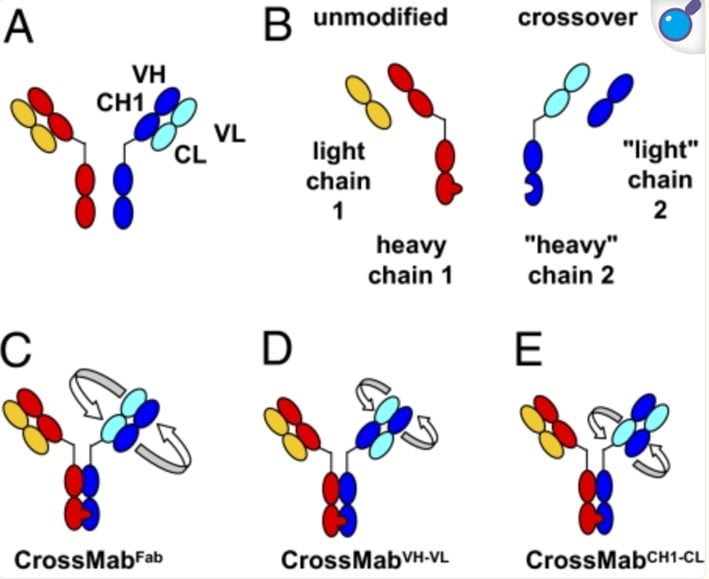
Fig. 1. Schematic diagram of the Fab domain exchange resulting in the generation of a
bispecific antibody when combined with the KiH technology. Dark colors indicate heavy-chain domains.
Light colors indicate light-chain domains. (A) Both arms of the intended bispecific antibody. (B) Design
of the four chains of the bispecific antibody. Heavy-chain heterodimerization is achieved by use of the
KiH technology. (C) Crossover of the complete VH-CH1 and VL-CL domains. (D and E) Crossover of only the
VH and VL domains (D) or the CH1 and CL domains (E) within the Fab region of one half of the bispecific
antibody. Doi:10.1073/pnas.1019002108
Since its inception, CrossMab technology has evolved over the past decade to become one of the most
mature and widely applied platforms in the field of bispecific antibodies. As of 2021, at least 19 bispecific
antibodies and fusion proteins developed using CrossMab technology have advanced into clinical trials.
Knob-Into-Hole (KiH)
Early antibody drugs were derived from mice, but due to strong immune reactions in humans, more
human-like antibodies have been developed. Based on their origin, therapeutic antibodies are classified into
four types:The Knob-Into-Hole (KiH) technology involves point mutations in the CH3 domain of antibodies, where
specific mutations are introduced into the heavy chains of two distinct antibodies. One heavy chain is
engineered with a "knob" mutation, typically replacing a specific amino acid residue with a bulkier one, such as
T366W. Conversely, the other heavy chain is modified with a "hole" mutation, replacing specific amino acid
residues with smaller ones, commonly T366S, L368A, Y407V [2, 3]. Through these modifications, the
correct
assembly of the heavy chains from two different antibodies is achieved, resulting in the formation of stable
bispecific antibodies. The KiH technology ensures proper pairing and stability of the antibody heavy chains,
thereby enhancing production efficiency and functional efficacy.
Format Feature
The crossover approach preserves the antigen-binding regions of the parental antibodies intact,
thereby offering a methodology to transform any given pair of antibodies—such as those with demonstrated
clinical efficacy—into a nearly natural bispecific IgG antibody. Irrespective of individual amino acid
sequences, the frameworks of the CH1 and CL domains are structurally analogous and can be readily superimposed.
Consequently, the CH1 domain can be entirely replaced with the CL domain, and vice versa, without compromising
the overall architecture of the Fab region. Since the Fc portion of these molecules remains unaltered,
Fc-mediated effector functions and properties are retained, including complement activation, neonatal Fc
receptor interactions, or FcγRIIIa interactions necessary for antibody-dependent cellular cytotoxicity (ADCC).
Although the domain exchange has been designed to induce correct pairing of the light chains with
their cognate heavy chains, their preparation is not completely free of side products. In the case of the
CrossMab(Fab), formation of a nonfunctional monovalent heavy-chain heterodimer (Fig.2F) and a nonfunctional Fab
fragment (a light-chain heterodimer; Fig.2G) can be expected. In the case of the CrossMab(VH-VL), the formation
of the antibody depicted in Fig.2I can be expected because light-chain variable domains tend to form Bence–Jones
homodimers[4, 5]. In the CrossMab(VH-VL), the VL domain of the unmodified light chain thus is able to
combine
with the VL domain of the domain-exchanged heavy chain. This tendency is amplified by the natural interaction
between the CL domain of the unmodified light chain and the CH1 domain of the heavy chain, thus leading to an
antibody containing two light chains. In the case of the CrossMab(CH1-CL), no side products with unwanted domain
association can be predicted from the analysis of chain compositions (Fig.2J). To further refine the byproduct
profile, optimization of the heavy chain to light chain ratio during both transient expression and stable cell
line generation may be considered[1].
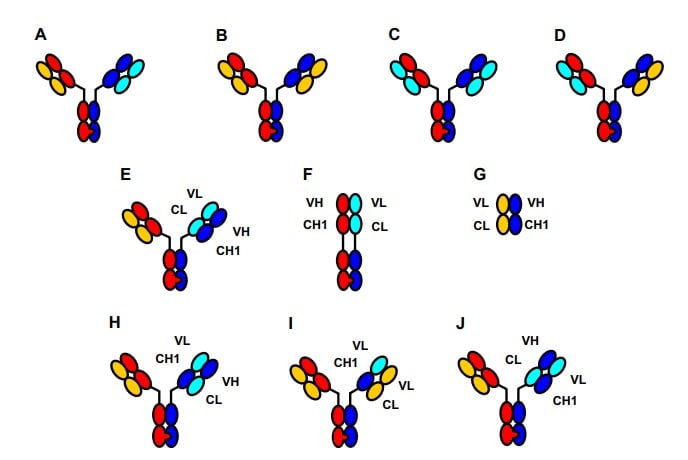
Fig. 2. Schematic representation of the antibodies discussed in the text. The desired
antibody (A) and undesired side products (B, C, and D) resulting from application of the
knobs-into-holes (KiH) technology. (E) Cross(MabFab). (F) Unspecific monovalent antibody and (G)
unspecific Fab observed as main side products in the generation of E. (H) CrossMab(VH-VL) and (I) its
major side product which results from Bence–Jones interaction of the wrong light chain with the
domain-exchanged heavy chain. (J) CrossMab(CH1-CL) as used in the in vivo experiments.
Doi:10.1073/pnas.1019002108
Due to their close resemblance to natural antibodies and the absence of additional non-native
antibody domains or linker sequences, asymmetric formats are likely to exhibit minimal immunogenicity. This
asymmetry also implies that bispecific antibodies with a conventional IgG structure are monovalent on each arm.
Compared to multivalent formats, the reduced binding affinity may compromise efficacy in certain therapeutic
applications. Furthermore, the incorporation of an Fc region can optimize pharmacokinetic profiles and augment
effector functions.
It is also noteworthy that many bispecific antibodies (BsAbs) utilizing CrossMab target CD3, and
nearly all CD3-targeting BsAbs currently in clinical development feature either engineered Fc regions or are
designed as Fc-lacking bispecific antibody fragments to minimize FcγR binding. This design choice is primarily
driven by the following considerations: 1) The Fc region of CD3 antibodies may bind to Fc receptors on other
immune cells or engage in off-target interactions with non-target cells, potentially triggering robust local
cytokine release and T cell-mediated hepatotoxicity [6]. 2) Fc-mediated effector mechanisms may
impede
tumor-specific T cell redirection and tumor cell killing [7]. These observations suggests that
CD3-targeting
bispecific T cell engagers (bsTCEs) require near-complete suppression of Fc-mediated effector functions to
minimize off-target toxicity and maximize therapeutic efficacy. Therefore, the optimal scenario is an antibody
Fc region that is highly inert, devoid of residual FcγR and C1q interaction capabilities, while retaining FcRn
binding capacity.
Antibody in this format
Vanucizumab
Vanucizumab (RG7221), one of the first bispecific antibodies employing CrossMab technology to enter
clinical trials, targets VEGF and Ang-2. Preclinical studies demonstrated potent anti-tumor and anti-angiogenic
activity across multiple models. In Phase I trials as monotherapy, vanucizumab exhibited favorable tolerability,
promising anti-tumor efficacy, low immunogenicity, and pharmacodynamic effects consistent with its intended
mechanism. However, Phase II evaluation in untreated metastatic colorectal cancer patients failed to demonstrate
significant clinical advantage compared to bevacizumab in combination with FOLFOX-6 chemotherapy, leading to
discontinuation of its development. Similarly, despite robust preclinical rationale, Phase Ib combination
studies with PD-L1 inhibitor atezolizumab (NCT01688206) and CD40 agonist selicrelumab (NCT02665416) were
ultimately terminated.
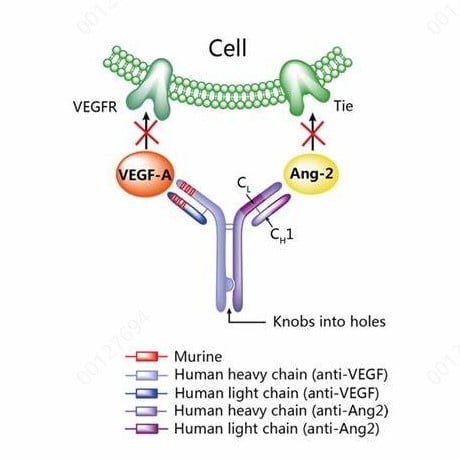
Fig. 3. Structural Schematic of Vanucizumab. Vanucizumab (RG7221) is a bispecific antibody
engineered using CrossMab technology, designed to simultaneously target vascular endothelial growth
factor A (VEGF-A) and angiopoietin-2 (Ang-2).
Faricimab
Faricimab (RO6867461, RG7716) is a heterodimeric 1+1 format bispecific antibody targeting VEGF and
Ang-2. By introducing the P329G LALA and 3A mutations into the IgG1 Fc region containing the Knob-Into-Hole
(KiH) modification, FcγR-mediated effector functions are effectively abolished while preserving FcRn recycling
under conditions of low systemic exposure. Recently, four independent pivotal Phase Ⅲ trials in patients with
wet age-related macular degeneration(wAMD) and diabetic macular edema(DME) reported positive outcomes, meeting
their primary endpoints (NCT03823287, NCT03823300, NCT03622580, NCT03622593). Based on these data, Faricimab has
been submitted for FDA review.
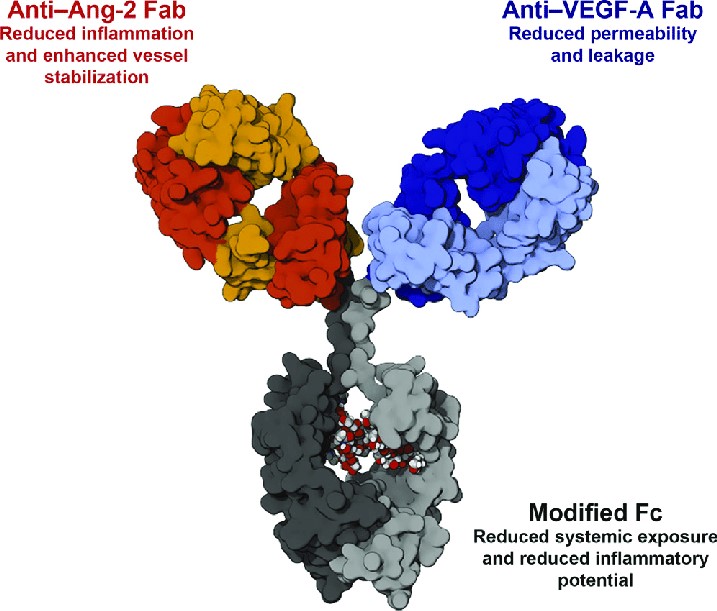
Fig. 4. Structural Schematic of Faricimab. Faricimab (RO6867461, RG7716) is a bispecific
antibody designed to simultaneously target vascular endothelial growth factor A (VEGF-A) and
angiopoietin-2 (Ang-2).
10E8.4/iMab
10E8.4/iMab (TMB-370), a bispecific antibody targeting HIV-1 and CD4 in a 1+1 format, demonstrated
remarkable prophylactic efficacy against high-dose intravenous HIV challenge. Notably, even after systemic
depletion of CD8α+ lymphocytes, no evidence of viral infection was observed over a five-week monitoring period.
In contrast, control animals developed plasma viremia within one week post high-dose viral inoculation. These
findings underscore the potential of 10E8.4/iMab (TMB-370) as a highly promising strategy for preventing
high-dose intravenous HIV transmission.
Iomvastoming
Iomvastoming (RG7769, PD1-TIM3) is a novel heterodimeric bispecific antibody in the 1+1 CrossMab
VH-VL format, targeting PD-1 and TIM-3. It features a high-affinity PD-1 Fab arm and a low-affinity TIM-3 Fab
arm, enabling selective targeting of PD-1+ and PD-1+TIM-3+ T cells while sparing PD-1-TIM-3+ myeloid and NK
cells. Compared to bivalent TIM-3 antibodies, RG7769 exhibits reduced binding to TIM-3+ myeloid and NK cells but
preferentially engages dysfunctional T cells expressing PD-1 or both PD-1 and TIM-3, such as tumor-infiltrating
lymphocytes (TILs) within the tumor microenvironment. Due to its monovalent design, RG7769 induces less antibody
internalization on activated T cells compared to bivalent TIM-3 antibodies, thereby overcoming a major
limitation of TIM-3 antibody-mediated cellular pooling [8].
Iomvastoming
Tobemstomig is a bispecific antibody in the 1+1 format targeting PD-1 and LAG-3. While PD-1
checkpoint inhibitors have demonstrated unprecedented clinical efficacy across multiple cancer indications, only
approximately 20-30% of patients achieve durable benefits from such therapies. One potential explanation for
this limited response is the compensatory upregulation or activation of alternative immune checkpoint pathways,
such as LAG-3, in tumor-reactive T cells. LAG-3 is constitutively expressed on regulatory T cells (Tregs), and
its blockade has been reported to enhance the suppressive function of Tregs, thereby potentially diminishing the
therapeutic efficacy of LAG-3 inhibition on tumor-reactive T cells. In other words, although blocking LAG-3 may
help activate tumor-reactive T cells, concurrent enhancement of Treg-mediated suppression could counteract the
overall therapeutic benefit. Tobemstomig addresses this challenge through its unique design: it exhibits a
20-fold higher affinity for PD-1 compared to LAG-3, enabling selective targeting of tumor-reactive T cells over
Tregs. Additionally, the antibody incorporates P329G LALA mutations to silence Fc-mediated effector functions,
reducing internalization and enhancing safety. In several murine models, Tobemstomig demonstrated superior tumor
control or eradication compared to combinations of single-specificity PD-1 and LAG-3 antibodies. Currently,
Tobemstomig has completed dose escalation and is undergoing Phase II clinical trials for several solid tumors
[9].
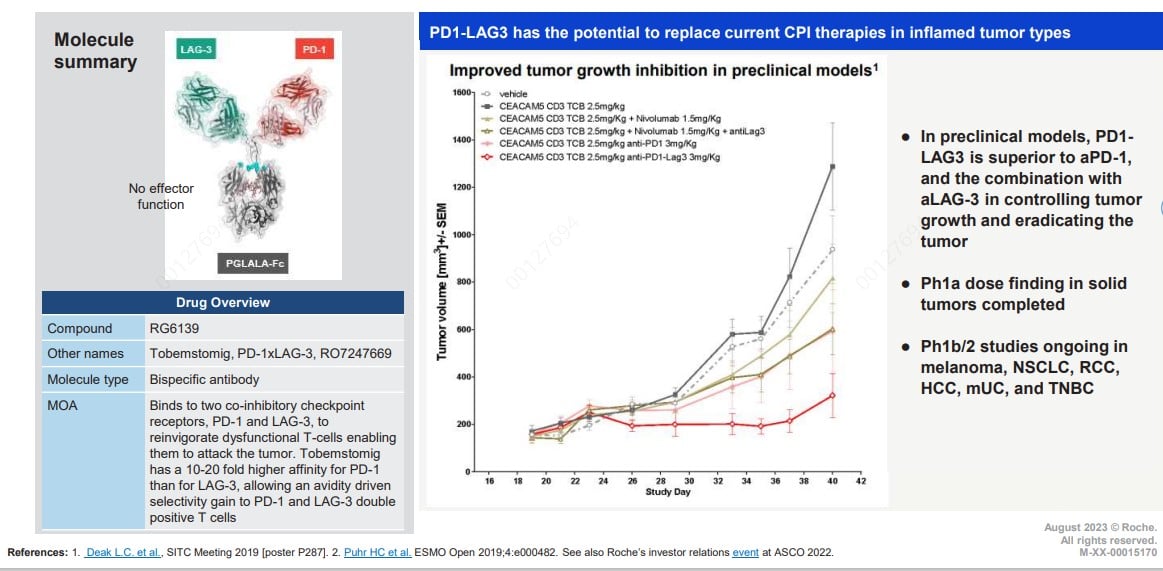
Fig. 5. Structural Schematic of Tobemstomig. Tobemstomig is a bispecific antibody designed
in the 1+1 format, targeting programmed cell death protein 1 (PD-1) and lymphocyte-activation gene 3
(LAG-3).
References
[1] Schaefer, W., Regula, J. T., Bähner, M., et al. (2011). Immunoglobulin domain crossover as a
generic approach for the production of bispecific IgG antibodies. Proceedings of the National Academy of
Sciences of the United States of America, 108(27), 11187-11192. https://doi.org/10.1073/pnas.1019002108
[2] Merchant, A. M., Zhu, Z., Yuan, J. Q., et al. (1998). An efficient route to human bispecific
IgG. Nature Biotechnology, 16(7), 677-681. https://doi.org/10.1038/nbt0798-677
[3] Ridgway, J. B., Presta, L. G., & Carter, P. (1996). 'Knobs-into-holes' engineering of antibody
CH3 domains for heavy chain heterodimerization. Protein Engineering, 9(7), 617-621.
https://doi.org/10.1093/protein/9.7.617 . PMID: 8844834.
[4] Gally, J. A., & Edelman, G. M. (1964). Protein-protein interactions among L-polypeptide chains
of Bence-Jones proteins and human gamma-globulins. Journal of Experimental Medicine, 119, 817–836.
[5] Schiffer, M., et al. (1970). A preliminary crystallographic investigation of human L-type
Bence-Jones protein. Journal of Biological Chemistry, 245, 728–730.
[6] Chatenoud, L., et al. (1990). In vivo cell activation following OKT3 administration. Systemic
cytokine release and modulation by corticosteroids. Transplantation, 49, 697–702.
[7] Labrijn, A. F., et al. (2017). Efficient generation of bispecific murine antibodies for
pre-clinical investigations in syngeneic rodent models. Scientific Reports, 7, 2476.
[8] RG7769 (PD1-TIM3), a novel heterodimeric avidity-driven T cell specific PD-1/TIM-3 bispecific
antibody lacking Fc-mediated effector functions for dual checkpoint inhibition to reactivate dysfunctional T
cells. Cancer Research, 80(16_Supplement), 2270 (2020).
[9] Tobemstomig, a novel bispecific checkpoint inhibitory antibody to preferentially block PD-1 and
LAG-3 on CD8 TILs over Tregs. Cancer Research, 84(6_Supplement), 7534 (2024).
 Most Popular Reagents
Most Popular Reagents
 Instruments
Instruments
 Antibodies
Antibodies
 Protein Electrophoresis and Blotting
Protein Electrophoresis and Blotting
 Molecular Biology
Molecular Biology
 Stable Cell Lines
Stable Cell Lines
 Cell Isolation and Activation
Cell Isolation and Activation
 IVD Raw Materials
IVD Raw Materials
 Therapy Applications
Therapy Applications
 Resources
Resources